15. Electronegativity Equalization Method#
This notebook shows how to compute atomic charges using the electronegativity equalization method. The basic idea is that dependence of the energy of an atom (or group) in a molecule can be written as a Taylor series,
which, by convention, we truncate at second order. The first derivative is the electronic chemical potential (the negative of the electronegativity, \(\chi\); also called the Fermi level) and the second derivative is the chemical hardness, \(\eta\) (also called the band gap). Thus, we can write
Using the known energies of the \(N_0\), \(N_0 + 1\), and \(N_0 - 1\) states, we can solve for \(\mu_A\) and \(\eta_A\), obtaining the Mulliken electronegativity and Parr-Pearson hardness,
where \(I = E(N_0) - E(N_0 + 1) \ge 0\) is the ionization potential and \(A = E(N_0 - 1) - E(N_0) \ge 0\) is the electron affinity.
The total energy is then,
This assumes that the atoms/groups “reference state” is neutral so that their charge is just \(q_A = -\Delta N_A\). The last term is the Coulomb interaction between the charges, where \(J_{AB}\) is the Coulomb integral between the charge distributions of atoms/groups \(A\) and \(B\). We could evaluate the Coulomb integral between the charge distributions analytically using AtomDB or Bfit, but this is not what is usually done in practice. Instead, the Coulomb interaction is usually screened, so that for electrostatic interactions between bonded atoms and atoms bonded to a common atom (linked by a bond angle) the Coulomb interaction is nearly neglected, while for atoms linked by a torsion the electrostatic interaction is screened. We’ll use an erfgau screening,
Now, to obtain the charges, we minimize the total energy with respect to the amount of electron transfer, \(\Delta N_A\), subject to the constraint that the total charge is fixed, \(\sum_A \Delta N_A = -Q\).
Forcing the constraint with the Lagrange multiplier, \(\mu_{\text{total}}\), we have the Lagrangian,
Differentiating the Lagrangian and setting it equal to zero gives a system of \(P+1\) linear equations, where \(P\) is the number of atoms/groups,
Using the explicit expression for the total energy, this system of equations can be written in matrix form as
where
We take benchmark values of the chemical potentials and hardnesses from this reference, using AtomDB.
!pip install qc-atomdb
!pip install qc-iodata
Collecting qc-atomdb
Downloading qc_AtomDB-0.0.2.post5-py3-none-any.whl.metadata (7.0 kB)
Requirement already satisfied: numpy>=1.16 in /opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages (from qc-atomdb) (2.4.4)
Requirement already satisfied: scipy>=1.4 in /opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages (from qc-atomdb) (1.17.1)
Collecting msgpack>=1.0.0 (from qc-atomdb)
Downloading msgpack-1.1.2-cp314-cp314-manylinux2014_x86_64.manylinux_2_17_x86_64.manylinux_2_28_x86_64.whl.metadata (8.1 kB)
Collecting msgpack-numpy>=0.4.8 (from qc-atomdb)
Downloading msgpack_numpy-0.4.8-py2.py3-none-any.whl.metadata (5.0 kB)
Collecting h5py>=3.6.0 (from qc-atomdb)
Downloading h5py-3.16.0-cp314-cp314-manylinux_2_28_x86_64.whl.metadata (3.0 kB)
Collecting importlib-resources>=3.0.0 (from qc-atomdb)
Downloading importlib_resources-6.5.2-py3-none-any.whl.metadata (3.9 kB)
Collecting pooch>=1.8.1 (from qc-atomdb)
Downloading pooch-1.9.0-py3-none-any.whl.metadata (10 kB)
Requirement already satisfied: platformdirs>=2.5.0 in /opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages (from pooch>=1.8.1->qc-atomdb) (4.9.4)
Requirement already satisfied: packaging>=20.0 in /opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages (from pooch>=1.8.1->qc-atomdb) (26.0)
Requirement already satisfied: requests>=2.19.0 in /opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages (from pooch>=1.8.1->qc-atomdb) (2.33.1)
Requirement already satisfied: charset_normalizer<4,>=2 in /opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages (from requests>=2.19.0->pooch>=1.8.1->qc-atomdb) (3.4.7)
Requirement already satisfied: idna<4,>=2.5 in /opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages (from requests>=2.19.0->pooch>=1.8.1->qc-atomdb) (3.11)
Requirement already satisfied: urllib3<3,>=1.26 in /opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages (from requests>=2.19.0->pooch>=1.8.1->qc-atomdb) (2.6.3)
Requirement already satisfied: certifi>=2023.5.7 in /opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages (from requests>=2.19.0->pooch>=1.8.1->qc-atomdb) (2026.2.25)
Downloading qc_AtomDB-0.0.2.post5-py3-none-any.whl (62.0 MB)
?25l ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 0.0/62.0 MB ? eta -:--:--
━━━━━╺━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 8.4/62.0 MB 61.8 MB/s eta 0:00:01
━━━━━━━━╺━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 12.6/62.0 MB 38.4 MB/s eta 0:00:02
━━━━━━━━━━╸━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 16.8/62.0 MB 29.0 MB/s eta 0:00:02
━━━━━━━━━━━━━━━━━━╸━━━━━━━━━━━━━━━━━━━━━ 29.1/62.0 MB 36.7 MB/s eta 0:00:01
━━━━━━━━━━━━━━━━━━━━━━━━━╺━━━━━━━━━━━━━━ 38.8/62.0 MB 39.2 MB/s eta 0:00:01
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╸━━━━ 55.6/62.0 MB 50.3 MB/s eta 0:00:01
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 62.0/62.0 MB 47.9 MB/s 0:00:01
?25hDownloading h5py-3.16.0-cp314-cp314-manylinux_2_28_x86_64.whl (5.4 MB)
?25l ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 0.0/5.4 MB ? eta -:--:--
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 5.4/5.4 MB 113.1 MB/s 0:00:00
?25hDownloading importlib_resources-6.5.2-py3-none-any.whl (37 kB)
Downloading msgpack-1.1.2-cp314-cp314-manylinux2014_x86_64.manylinux_2_17_x86_64.manylinux_2_28_x86_64.whl (419 kB)
Downloading msgpack_numpy-0.4.8-py2.py3-none-any.whl (6.9 kB)
Downloading pooch-1.9.0-py3-none-any.whl (67 kB)
Installing collected packages: msgpack, importlib-resources, h5py, pooch, msgpack-numpy, qc-atomdb
?25l
━━━━━━━━━━━━━╺━━━━━━━━━━━━━━━━━━━━━━━━━━ 2/6 [h5py]
━━━━━━━━━━━━━━━━━━━━╺━━━━━━━━━━━━━━━━━━━ 3/6 [pooch]
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╺━━━━━━ 5/6 [qc-atomdb]
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╺━━━━━━ 5/6 [qc-atomdb]
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 6/6 [qc-atomdb]
?25h
Successfully installed h5py-3.16.0 importlib-resources-6.5.2 msgpack-1.1.2 msgpack-numpy-0.4.8 pooch-1.9.0 qc-atomdb-0.0.2.post5
Collecting qc-iodata
Downloading qc_iodata-1.0.1-py3-none-any.whl.metadata (4.4 kB)
Requirement already satisfied: numpy>=1.26.4 in /opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages (from qc-iodata) (2.4.4)
Requirement already satisfied: scipy>=1.13.1 in /opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages (from qc-iodata) (1.17.1)
Requirement already satisfied: attrs>=21.3.0 in /opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages (from qc-iodata) (26.1.0)
Downloading qc_iodata-1.0.1-py3-none-any.whl (3.4 MB)
?25l ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 0.0/3.4 MB ? eta -:--:--
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 3.4/3.4 MB 28.1 MB/s 0:00:00
?25h
Installing collected packages: qc-iodata
Successfully installed qc-iodata-1.0.1
import numpy as np
from scipy.special import erf
def erfgau(r, alpha=0.5):
"""
Savin's erfgau long-range interaction:
v(r) = erf(mu*r)/r - (2*mu/sqrt(pi)) * exp(-(mu*r)^2 / 3)
Parameters
----------
r
Interparticle distance(s). May be a positive float or a NumPy array
of positive distances.
alpha
Range-separation parameter. Must be nonnegative. The default
value is set so that for a 1-3 interactions of ~6 bohr the erfgau
screening is roughly 20%. As this number increases the screening becomes
less aggressive.
Returns
-------
float or np.ndarray
The erfgau interaction evaluated at r.
"""
if alpha < 0.0:
raise ValueError("alpha must be nonnegative.")
# Set a tolerance for zero to avoid issues with very small distances
tol = 1e-12
# Treat scalar case separately
if np.isscalar(r):
r = float(r)
if r < 0:
raise ValueError("r must be nonnegative.")
elif r < tol:
# For very small r, use the limit as r -> 0 to avoid numerical issues
return 0.0
else:
x = alpha * r
return erf(x) / r - (2.0 * alpha / np.sqrt(np.pi)) * np.exp(-(x**2) / 3.0)
r_arr = np.asarray(r, dtype=float)
if np.any(r_arr < 0.0):
raise ValueError("All elements of r must be nonnegative.")
x = alpha * r_arr
# Form an array to hold the input and initalize it to zero
erfgau = np.zeros_like(r_arr)
# For very small r, zero is the answer so the intialization is correct.
# For larger r, compute the erfgau interaction as normal. We can do this with
# np.where.
erfgau = np.where(r_arr < tol, 0.0, erf(x) / r_arr - (2.0 * alpha / np.sqrt(np.pi)) * np.exp(-(x**2) / 3.0))
return erfgau
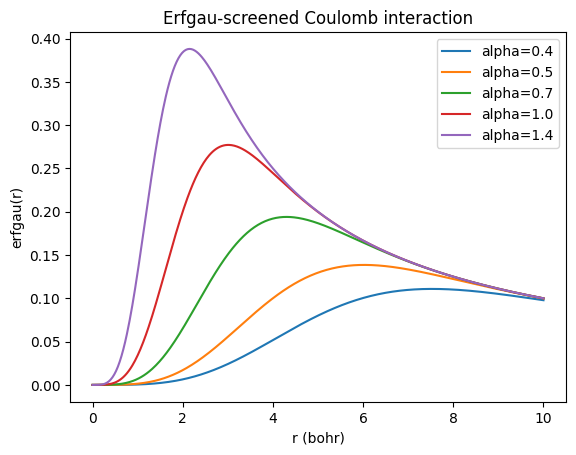
#Plot Erfgau vs r for different parameters.
import matplotlib.pyplot as plt
r = np.linspace(0, 10, 500)
for alpha in [0.4, 0.5, 0.7, 1.0, 1.4]:
plt.plot(r, erfgau(r, alpha), label=f"alpha={alpha}")
plt.xlabel("r (bohr)")
plt.ylabel("erfgau(r)")
plt.title("Erfgau-screened Coulomb interaction")
plt.legend()
plt.show()
/tmp/ipykernel_2894/1849752753.py:55: RuntimeWarning: invalid value encountered in divide
erfgau = np.where(r_arr < tol, 0.0, erf(x) / r_arr - (2.0 * alpha / np.sqrt(np.pi)) * np.exp(-(x**2) / 3.0))
# Load the geometry from a file (using IOData)
from urllib.request import urlretrieve
from iodata import load_one
url = "https://github.com/theochem/chemtools/raw/refs/heads/master/chemtools/data/examples/dichloropyridine26_q+0.fchk"
urlretrieve(url, "dichloropyridine26_q0.fchk")
mol0 = load_one("dichloropyridine26_q0.fchk")
print(mol0.atnums)
print(mol0.atcoords)
[17 17 7 6 6 6 6 6 1 1 1]
[[-4.95130924e+00 2.31173977e+00 -2.64561659e-04]
[ 4.95149821e+00 2.31139962e+00 2.07869875e-04]
[ 7.55890453e-05 1.84250188e+00 -1.88972613e-05]
[-9.44863066e-05 -3.41775868e+00 1.13383568e-04]
[-2.28277027e+00 -2.09580077e+00 -1.88972613e-05]
[ 2.28267578e+00 -2.09595195e+00 1.70075352e-04]
[-2.13763930e+00 5.39856962e-01 -3.77945227e-05]
[ 2.13769600e+00 5.39705784e-01 7.55890453e-05]
[-1.70075352e-04 -5.47831606e+00 1.51178091e-04]
[-4.10063012e+00 -3.05289036e+00 -7.55890453e-05]
[ 4.10047894e+00 -3.05315492e+00 2.83458920e-04]]
# Retrieve the chemical potentials and hardnesses using AtomDB
import atomdb
from atomdb import Element
def nist_mu_eta_for_atoms(atnums):
"""return a numpy array of mu's and eta's for the given atomic numbers"""
atnums = np.asarray(atnums, dtype=int)
mu = np.empty(len(atnums))
eta = np.empty(len(atnums))
for i, z in enumerate(atnums):
mult = Element(z).mult # neutral ground-state multiplicity
sp = atomdb.load(z, charge=0, mult=mult, dataset="nist")
mu[i] = sp.mu
eta[i] = sp.eta
return mu, eta
# Example with your molecule atomic numbers:
# atnums = h2o.atnums # or your molecule's atnums
mu, eta = nist_mu_eta_for_atoms(mol0.atnums)
print("mu =", mu)
print("eta =", eta)
Downloading data from 'https://raw.githubusercontent.com/theochem/AtomDBdata/main/nist/db/Cl_000_002_000.msg' to file '/opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages/atomdb/datasets/nist/db/Cl_000_002_000.msg'.
SHA256 hash of downloaded file: 07a26c7400ba83f656824316604b3fae3916aaad869d4ac8cf86a3980dae5380
Use this value as the 'known_hash' argument of 'pooch.retrieve' to ensure that the file hasn't changed if it is downloaded again in the future.
Downloading data from 'https://raw.githubusercontent.com/theochem/AtomDBdata/main/nist/db/N_000_004_000.msg' to file '/opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages/atomdb/datasets/nist/db/N_000_004_000.msg'.
SHA256 hash of downloaded file: be4cbc4099c0e301ea914ce7c558555ad38ccee7425b57bf93c94e94f44065ae
Use this value as the 'known_hash' argument of 'pooch.retrieve' to ensure that the file hasn't changed if it is downloaded again in the future.
Downloading data from 'https://raw.githubusercontent.com/theochem/AtomDBdata/main/nist/db/C_000_003_000.msg' to file '/opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages/atomdb/datasets/nist/db/C_000_003_000.msg'.
SHA256 hash of downloaded file: ead0c0345a38ceee1b301266de10c9571fdaed2e711a0220b4479920bdd9ae2b
Use this value as the 'known_hash' argument of 'pooch.retrieve' to ensure that the file hasn't changed if it is downloaded again in the future.
Downloading data from 'https://raw.githubusercontent.com/theochem/AtomDBdata/main/nist/db/H_000_002_000.msg' to file '/opt/hostedtoolcache/Python/3.14.3/x64/lib/python3.14/site-packages/atomdb/datasets/nist/db/H_000_002_000.msg'.
SHA256 hash of downloaded file: 6ac81ca278c2b1e98af89d2ea9e018c0766fbd041d2e588e0e34d2ab620a8867
Use this value as the 'known_hash' argument of 'pooch.retrieve' to ensure that the file hasn't changed if it is downloaded again in the future.
mu = [-0.30465188 -0.30465188 -0.26716757 -0.23005076 -0.23005076 -0.23005076
-0.23005076 -0.23005076 -0.26386013 -0.26386013 -0.26386013]
eta = [0.34360616 0.34360616 0.53396765 0.36749322 0.36749322 0.36749322
0.36749322 0.36749322 0.4718613 0.4718613 0.4718613 ]
# Set up the equations to solve for the charges and print out the charges.
from scipy.spatial.distance import pdist, squareform
def eem_charges(atnums, atcoords, mu, eta, mol_charge, alpha=0.5):
"""Compute EEM atomic charges.
Builds and solves the (P+1) x (P+1) EEM linear system A·δ = m, where
the diagonal contains the atomic hardnesses, the off-diagonal atom-atom
blocks contain the erfgau-screened Coulomb interaction J_AB, the border
row and column enforce the charge constraint, and the right-hand side
contains the negative chemical potentials and the total molecular charge.
Parameters
----------
atnums : array-like of int, shape (P,)
Atomic numbers (used only to determine P; properties come from mu/eta).
atcoords : numpy.ndarray, shape (P, 3)
Atomic coordinates in bohr.
mu : numpy.ndarray, shape (P,)
Atomic chemical potentials (negative electronegativities).
eta : numpy.ndarray, shape (P,)
Atomic chemical hardnesses.
mol_charge : float
Total molecular charge Q.
alpha : float
Range-separation parameter for the erfgau Coulomb screening.
Returns
-------
charges : numpy.ndarray, shape (P,)
EEM atomic charges q_A = -Delta N_A.
mu_total : float
Equilibrated chemical potential (Lagrange multiplier).
"""
atcoords = np.asarray(atcoords)
mu = np.asarray(mu)
eta = np.asarray(eta)
P = len(mu)
# Build the (P+1) x (P+1) EEM matrix A
A = np.zeros((P + 1, P + 1))
# Off-diagonal atom-atom blocks: erfgau-screened Coulomb interaction.
# pdist returns only the upper-triangle distances (no zeros), so erfgau
# never sees r=0 and no divide-by-zero warning is raised.
A[:P, :P] = squareform(erfgau(pdist(atcoords), alpha))
# Diagonal: atomic hardnesses (set after the off-diagonal fill so they
# are not overwritten by the squareform step above).
np.fill_diagonal(A[:P, :P], eta)
# Border row and column: charge constraint
A[:P, P] = 1.0
A[P, :P] = 1.0
# A[P, P] = 0 (already zero from initialization)
# Build the right-hand side m
m = np.empty(P + 1)
m[:P] = -1*mu
m[P] = -1*mol_charge
# Solve A·delta = m where delta = [Delta_N_1, ..., Delta_N_P, mu_total]
delta = np.linalg.solve(A, m)
charges = -1*delta[:P] # q_A = -Delta N_A
mu_total = -1*delta[P]
return charges, mu_total
charges, mu_total = eem_charges(mol0.atnums, mol0.atcoords, mu, eta, mol_charge=0,alpha=0.5)
print("Atomic charges:", charges)
print("Equilibrated chemical potential:", mu_total)
print("Sum of charges:", charges.sum())
Atomic charges: [-0.28375011 -0.28374982 -0.01416517 0.18020443 0.1505785 0.15057809
0.06419838 0.0641997 -0.00754719 -0.01027312 -0.0102737 ]
Equilibrated chemical potential: -0.24684627271641874
Sum of charges: 1.0061396160665481e-16
Play around with the notebook and see how the results change when you adjust the screening parameter for the EEM Coulomb potential and/or the total molecular charge.